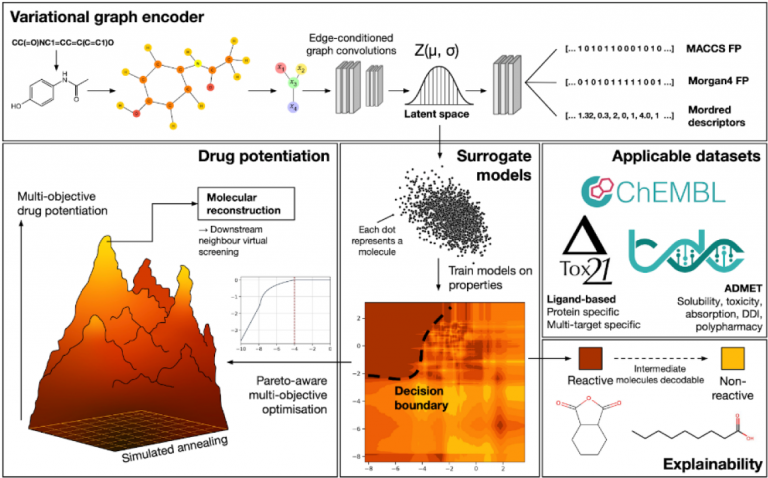
Molecules and compounds used in our daily drugs vary in shapes and sizes. Currently, there are an estimated 1063 unique compounds that have the potential to be used as pharmaceutical drugs. However, these molecules are discrete in nature and cannot be easily represented mathematically.
In this research, the researchers employed a variational graph encoder to convert individual molecules into a continuous numerical space. Consequently, each molecule can now be represented using 64 numbers.
Using these 64 numbers, they demonstrated the capability to predict various compound properties, including absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles.
This research specifically showcased that building smaller models based on these 64 numbers generated competent models that were comparable to or on par with state-of-the-art approaches, hence it was tagged as a “generalist” model. Additionally, the researchers discovered that the 64 numbers output from the model can be directly explored using other AI methods, enabling the effective specification of desired drug properties and automatic exploration for molecules that meet the criteria through the use of AI.
Molecule clustering from the ZINC database: Approximately 690 million molecules sourced from the ZINC database were split on their tranches and used for downstream training of the variational graph encoder.
Latent space surrogate model: Training of a specific machine learning model to predict the binding affinity of small molecules to a specific protein.
HPC resources: The project was allocated 500,000 CPU and 500,000 GPU hours from NSCC Singapore. These resources facilitated large-scale virtual screening, computational tasks and scoring of the test targets.
Increased Speed: The surrogate models developed by the researchers using latent space information, not only excel in predicting properties like ADMET but also notably accelerate virtual screening in ligand-based drug design.
Increased Accuracy: Employing the surrogate support vector machine model alongside five scoring functions, the researchers predicted compound affinity for five target proteins. The results demonstrated comparable accuracy in ranking ability to specialized scoring functions, while reducing computational time by 1-2 orders of magnitude. This significantly minimizes the computational resources required to process the evaluations.
Prospects: The NTU team will be working towards applying this innovative methodology to conduct virtual screenings across extensive datasets within the molecule library.






